大连化物所发现化学反应中新的量子干涉效应
近日,中国科学院大连化学物理研究所研究员肖春雷、孙志刚、中科院院士张东辉和杨学明团队在最简单化学反应氢原子加氢分子的同位素(H+HD→H2+D)反应中,发现了化学反应中新的量子干涉效应,有助于更深入地理解化学反应过程,丰富对化学反应的认识。
在化学反应中,量子干涉现象是普遍存在的。但是,想要准确理解这些干涉产生的根源非常困难,因为这些干涉的图样复杂,而且在实验上也难以精确分辨这些干涉图样的特征。
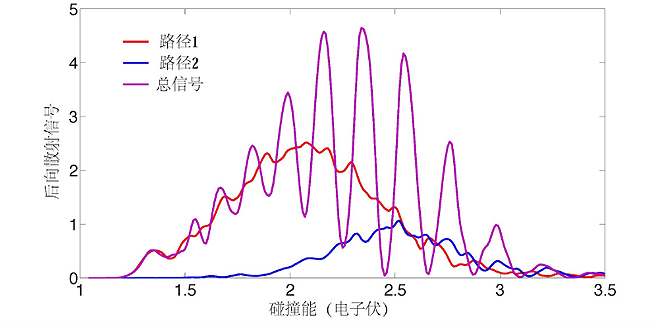
H+H2及其同位素的反应,是所有化学反应中最简单的。该体系只涉及三个电子,因此比较容易精确计算出这三个原子在不同构型时的相互作用力。在此基础上,通过求解相应的描述化学反应过程的薛定谔方程,就能够实现分子反应动力学过程的计算机模拟,从而做到在微观层次上深入理解化学反应过程。研究团队在2019年先期理论研究工作中发现,在特定散射角度上,H+HD反应生成的产物H2的多少会随碰撞能而呈现特别有规律的振荡。
针对这个有规律的振荡现象,该团队开展了理论结合实验的详细研究。实验上,通过改进了的交叉分子束装置,实现了在较高碰撞能处对后向散射(散射角度为180度)信号的精确测量。理论上,进一步发展了量子反应散射理论,创造性地发展了利用拓扑学原理来分析化学反应发生途径的方法。
拓扑学分析表明,这些后向散射的振荡实际上是由两条反应途径的干涉造成的。这两条反应途径对于后向散射均有显著贡献,但它们各自的幅度随着碰撞能变化并无显著变化,呈现缓慢的变化趋势。它们的相位随着碰撞能变化,一个呈线性增加,另外一个呈线性减小,因此,相互干涉的结果就呈现了强烈的有规律的振荡现象。
进一步采用经典轨线理论分析表明,其中一条反应途径对应于通常所熟知的直接反应过程,如下图G至I所示。而另外一条反应途径对应于一条类似于roaming机理的反应过程,如图A至F所示。由于这两条反应途径,刚好以相反的方向围绕于H+HD反应势能面上的锥形交叉,所以它们的干涉图样必须采用非绝热耦合的势能面来模拟计算才可以,这也体现了这个体系反应过程中的几何相位效应。尤其有趣的是,在所研究的碰撞能范围,通过漫游机理而发生的反应只占全部反应性的0.3%左右。而如此微弱的小部分反应性,能够清晰地被理论和实验所揭示出来。
该项研究一方面再次揭示了原子分子因碰撞而发生化学反应过程的量子性,另一方面也揭示了化学反应的途径是复杂的。尽管如此,简单的体系也仍然存在科学家们认识不到的事实。
相关文章于5月15日发表在《科学》(Science)杂志上。该研究得到国家自然科学基金项目和中科院战略性先导科技专项(B)等的支持。
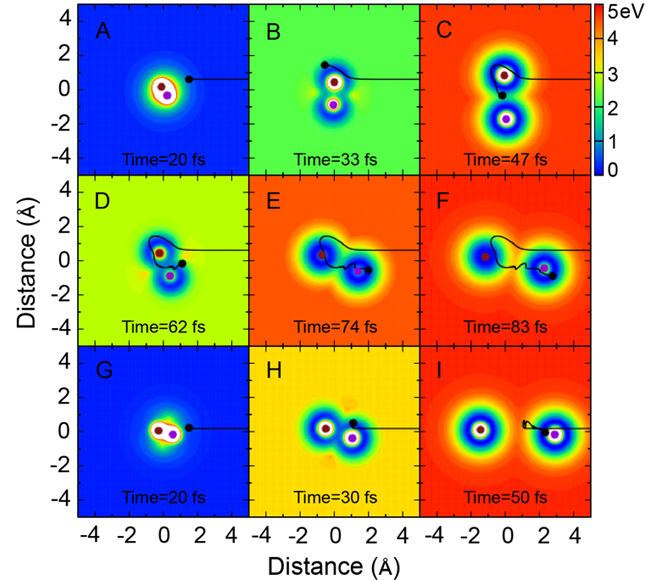
笛卡尔坐标系下,H+HD→H2+D反应的示意图
